


..就来聊聊它——分散剂。
在上一篇文章中,我们聊到了偶联剂。评论区有朋友提问:“这东西是不是跟分散剂差不多?”
希望看完这篇文章后,你能自己回答出:“分散剂和偶联剂到底有什么区别?”
如果你把纳米粒子、颜料粒子、填料颗粒倒进溶液里,搅一搅,放在那儿一晚上——第二天你会发现它们神奇地“抱成一团”了,有的还沉到瓶底。这是为什么?
答案其实藏在自然界..核心的一条规则里:一切系统都倾向于使自身能量..。
颗粒聚在一起,正是因为这样做能让系统能量变低,这主要表现在以下几种作用力:
无论你是高分子、无机粉体还是有机颜料,只要是分子或原子之间接近,一定会有范德华力。这种力不大,但胜在范围广、数量多,尤其在纳米尺度下,粒子间距离非常小,范德华力就“占主导了”。
只要颗粒靠近,它们就会互相吸引。
单个颗粒分散在液体中,意味着每个颗粒暴露在液体里,拥有较大的比表面积。而表面能是随面积升高而增加的,为了降低系统总能量,颗粒会选择聚在一起,减少暴露面积。
聚在一起能减少“接触面积”,降低表面能,系统就更稳定。
当然,不同体系下还会叠加其他因素,比如静电吸引、氢键作用、水合壳层等等,但本质还是在能量和热力学平衡上打转。
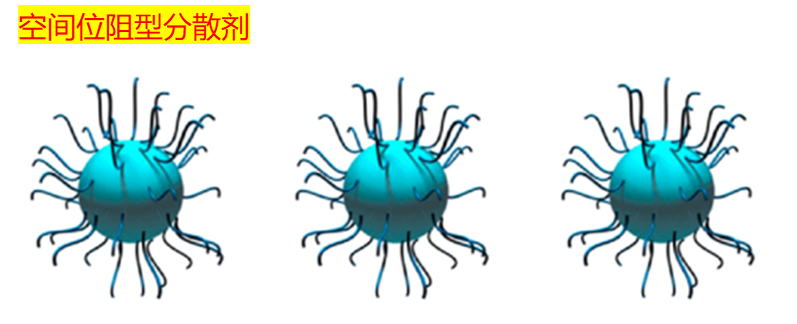
这类分散剂靠“占位置”来阻止颗粒靠近。
它们通常有两部分:
一端(锚定基团)牢牢吸附在颗粒表面;
另一端是高分子链段,在溶液中伸展开,像刺猬一样形成保护层。
当两个颗粒靠太近时,这些链段会重叠、压缩,导致系统熵降低(分子变得太规整了,不自然)。出于“本能”,系统会反弹回去,形成排斥力。
微观上说,这是链段热运动和电子云排斥共同作用的结果。
空间位阻型分散剂通过链段“挤开”机制,阻止颗粒靠近,从而实现分散。
这类分散剂让颗粒带上相同的电荷,比如都带负电。同性相斥,颗粒就不容易靠近。

..直观的例子就是两个带电小球会互相弹开,这背后就是库仑斥力。
从理论角度来看,DLVO理论认为颗粒间的作用力=吸引力(范德华力)+斥力(电荷斥力)。只要静电斥力足够大,就能抵消吸引,颗粒就能稳定分散。
电子层面上,表面电荷越多,双电层越厚,电位差越大,排斥力也越强。
静电型分散剂通过让颗粒同电排斥,维持分散稳定。
有些分散剂聪明一点,两个机制都用,比如一边挂着疏水链“撑体积”,一边还能解离出离子“带电荷”——这类我们通常称为复合型分散剂,在复杂体系里表现更稳定。
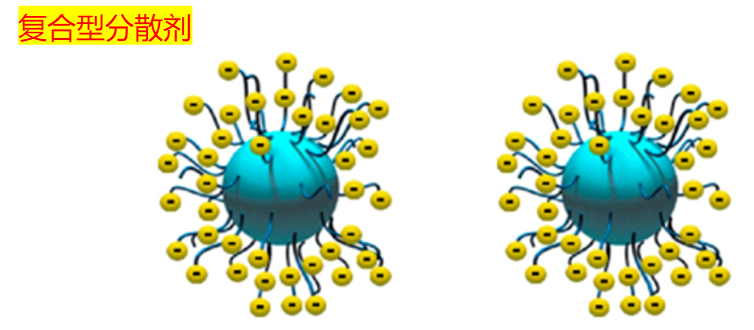
但再多类型的分散剂,其作用机理都可以归类为“空间阻隔”和“静电排斥”两种,或它们的组合。
如何选用适合自己的分散剂?
不同颗粒表面极性不同,能不能“锚定”住,取决于分散剂分子头部的官能团设计:
极性颗粒(如TiO₂、CaCO₃、SiO₂):表面通常含有羟基或离子键位点,推荐使用带羧基、羟基、磷酸基等极性锚定基团的分散剂,能通过氢键、配位键等方式吸附牢靠。
非极性颗粒(如炭黑、有机颜料):表面疏水光滑,需通过烷基链、芳香基团等疏水性基团靠范德华力吸附。
典型错误:只看润湿力而忽视吸附力,锚不住颗粒的“头”,再强的稳定链也白搭。
这是..基础但..容易忽视的一点,介质不同,稳定机制就变:
水性体系:颗粒易带电,可利用分散剂的离子基团(如羧酸、磺酸)提供电荷斥力,形成静电稳定屏障。聚电解质型(如PAA、PESNa)是主力。
油性体系:粒子间不易形成电荷,必须依赖高分子链段在表面形成空间障碍,防止近距离聚集,因此高分子型、嵌段共聚物型更可靠。
小结:水中靠“电”推开,油中靠“链”撑开。
很多体系不是静态的,真正决定分散剂能不能“长期在线”的,是它的耐工艺性能:
高剪切场合(球磨、砂磨、喷涂):容易剪断小分子或弱吸附分散剂,选用锚定更强、链段更粗壮的高分子型或嵌段型。
pH波动较大(如电泳、硅溶胶):静电型分散剂(PAA等)在中性偏酸条件下容易离子化能力下降或反应析出,稳定性失效。
高温/长储存体系:热会加速分子运动、解吸附,建议选用吸附强、结构稳定的复合型或高分子型分散剂。
提示:如果你用的体系有“砂磨+常温贮存+水性pH6~8”,请格外关注分散剂结构是否稳定!
这是很多实验失败的根源!静电稳定机制极易被电解质破坏:
在含盐体系中,自由离子会中和颗粒表面的电荷,形成“电荷屏蔽”效应,导致原本的电斥力减弱甚至消失,颗粒重新聚集。
聚电解质分散剂(如PAA、磺化物)在这种环境下稳定性下降明显,甚至会因结构变性而析出。
建议改用空间位阻型分散剂(如Solsperse 系列、Hypermer 系列),它们稳定性更依赖链段结构,对离子强度不敏感。
工业场景中常见电解质来源:助剂中残留Na⁺、Ca²⁺,水源硬度、添加电解质调pH 等。
 当前位置:
当前位置:
 微信
微信







